
Ehlers-Danlos综合征是一组疾病的影响结缔组织的组织支持皮肤,骨骼,血管和许多其他器官和组织。结缔组织中的缺陷导致这些症状的体征和症状,从轻度松弛的关节到危及生命的并发症。
各种形式的Ehlers-Danlos综合征已经以几种不同的方式分类。最初,11种形式的Ehlers-Danlos综合征用罗马数字命名,以表示类型(I型,II型等)。1997年,研究人员提出了一种更简单的分类(Villefranche命名法),将类型数量减少到6种,并根据其主要特征给出了描述性名称。2017年,分类更新,包括最近发现的罕见的Ehlers-Danlos综合症。2017年分类描述了13种Ehlers-Danlos综合征。
一种异常大范围的关节运动(运动过度)出现在大多数形式的Ehlers-Danlos综合症中,它是超移动型的标志性特征。患有过度活动的婴儿和儿童通常具有较弱的肌张力(肌张力减退),这可以延迟运动技能的发展,例如坐,站立和行走。松动的关节不稳定,易于脱位和慢性疼痛。在关节松弛症类型的Ehlers-Danlos综合征中,婴儿出生时臀部过度活动和脱臼。
许多患有Ehlers-Danlos综合症的人都有柔软,天鹅绒般的皮肤,具有高弹性(弹性)和脆弱性。受影响的人容易瘀伤,并且某些类型的病症也会导致异常瘢痕形成。具有Ehlers-Danlos综合征的经典形式的人经历伤口分裂开放,几乎没有出血并留下随时间变宽的疤痕以产生特有的“卷烟纸”疤痕。该病症的皮肤痘痘类型的特征在于皮肤松弛,下垂和皱纹,并且可能存在额外的(多余的)皮肤皱褶。
某些形式的Ehlers-Danlos综合征,特别是血管类型,以及较小程度上的kyphoscoliotic,经典和古典类型,可导致不可预测的血管撕裂(破裂),导致内部出血和其他可能危及生命的并发症。Ehlers-Danlos综合征的血管类型也与器官破裂的风险增加相关,包括在怀孕期间撕裂肠和破坏子宫。
其他类型的Ehlers-Danlos综合征有其他症状和体征。心脏瓣膜类型导致控制血液通过心脏运动的瓣膜的严重问题。患有kyphoscoliotic类型的人经历严重的脊柱弯曲随着时间的推移会恶化并且可以通过限制肺部扩张来干扰呼吸。一种称为脆性角膜综合征的Ehlers-Danlos综合征的特征在于眼睛(角膜)的清晰覆盖物和其他眼睛异常的薄度。脊柱增生异常型具有身材矮小和骨骼异常,例如异常弯曲(弯曲)的肢体。肌肉异常,包括肌张力减退和永久性弯曲关节(挛缩),是Ehlers-Danlos综合征的肌肉收缩和肌病形式的特征性征兆。牙周类型导致牙齿和牙龈的异常。
Ehlers-Danlos综合征发病率
所有类型的Ehlers-Danlos综合征的综合患病率似乎在全世界5,000个人中至少有1个。超移动和经典形式是最常见的; 超级移动类型可能影响多达1到5,000到20,000人,而经典类型可能在20,000到40,000人中有1个。其他形式的Ehlers-Danlos综合征很罕见,通常只有少数病例或患病家属在医学文献中描述。
Ehlers-Danlos综合征发病原因
已发现至少19个基因的突变导致Ehlers-Danlos综合征。COL5A1或COL5A2基因突变,或COL1A1基因中很少发生突变,可引起经典类型。TNXB基因的突变引起类似经典的类型,并且已经报道了极小比例的超移动型病例(尽管在这种类型的大多数人中,原因未知)。心脏瓣膜型和关节松弛型的一些病例是由COL1A2基因突变引起的; COL1A1基因突变也在关节松弛型患者中发现。大多数血管类型的病例是由COL3A1突变引起的基因,虽然很少这种类型是由某些COL1A1基因突变引起的。dermatosparaxis类型是由ADAMTS2基因的突变引起的。PLOD1或FKBP14基因突变导致kyphoscoliotic类型。其他罕见形式的Ehlers-Danlos综合征是由其他基因突变引起的。
与Ehlers-Danlos综合征相关的一些基因,包括COL1A1,COL1A2,COL3A1,COL5A1和COL5A2,提供了制造几种不同类型胶原蛋白的说明。这些件组装而形成成熟的胶原蛋白分子,其赋予结构和强度,以结缔组织的组织遍布全身。其他基因,包括ADAMTS2,FKBP14,PLOD1和TNXB,提供了制备处理,折叠或与胶原相互作用的蛋白质的说明。任何这些基因的突变都会破坏胶原蛋白的产生或加工,从而阻止这些分子正确组装。这些变化削弱了皮肤,骨骼和身体其他部位的结缔组织,从而产生了Ehlers-Danlos综合征的特征。
与最近描述的Ehlers-Danlos综合征类型相关的一些基因具有似乎与胶原无关的功能。对于许多这些基因,尚不清楚突变如何导致过度活动,弹性皮肤和这些病症的其他特征。
Ehlers-Danlos综合征遗传方式
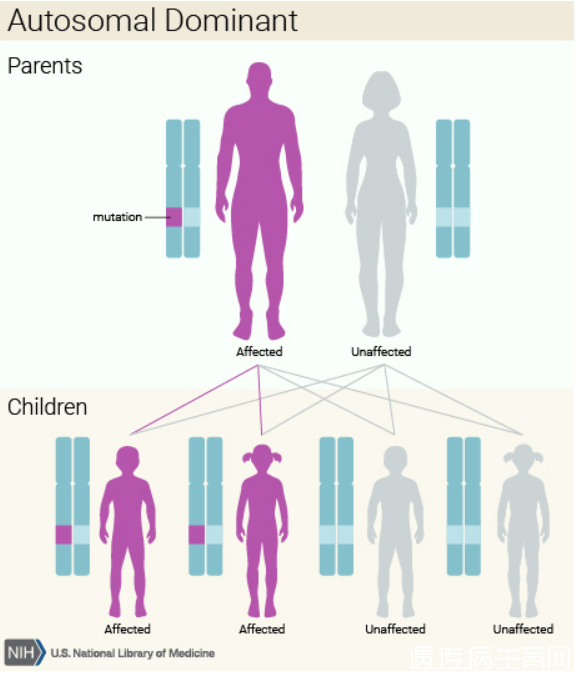
Ehlers-Danlos综合征的遗传模式因类型而异。这种疾病的典型,血管,关节紊乱和牙周形式,以及可能是超移动型,具有常染色体显性遗传模式。常染色体显性遗传意味着每个细胞中一个改变基因的拷贝足以引起疾病。在某些情况下,受影响的人从一个受影响的父母那里继承突变。其他病例来自新的(从头)基因突变 并且发生在没有家族疾病史的人群中。
Ehlers-Danlos综合征以及脆性角膜综合征的经典样,心脏瓣膜,皮肤吻合,脊柱后生,脊椎增生异常和肌肉骨折类型均以常染色体隐性遗传模式遗传。。在常染色体隐性遗传中,每个细胞中的两个基因拷贝被改变。大多数情况下,患有常染色体隐性遗传病的个体的父母是改变基因的一个拷贝的携带者,但是没有显示该疾病的体征和症状。
Ehlers-Danlos综合征的肌病类型可以具有常染色体显性遗传或常染色体隐性遗传模式。

