
苯丙酮尿症由挪威化学家Folling于1934年首先报道,因发现尿液中含苯丙酮酸而命名为苯丙酮尿症(phenylketonuria,PKU),又称高苯丙氨酸血症(hyperphenylalaninemia)。
苯丙酮尿症是一种罕见的代谢紊乱,影响人体分解蛋白质的方式。如果不在出生后不久即可治愈,PKU可能会破坏神经系统,造成智力障碍。PKU是由染色体12上的基因突变引起的。该基因编码称为PAH(苯丙氨酸羟化酶)的蛋白质,是肝脏中的一种酶。这种酶将氨基酸苯丙氨酸分解成人体所需的其他产品。当该基因发生突变时,PAH酶的形状发生改变,不能正确分解苯丙氨酸。苯丙氨酸累积在血液和毒素中的神经细胞(神经元)。
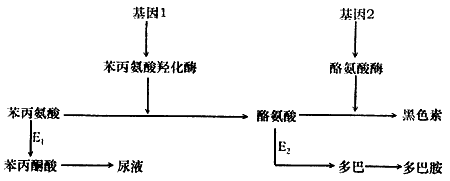
PKU是一种以智力低下为特征的先天性代谢缺陷,分经典型和非经典型两类。经典型是由于苯丙氨酸羟化酶(phenylalaninehydroxylase,PAH)缺乏所致。非经典型是由于生物喋呤代谢缺陷所引起,包括二氢生物喋啶还原酶(dihydropteridinereductase,DHPR)缺乏,或因生物喋呤合成相关的两种酶GTP环水解酶(GTPcyclohydrolase,GTP-CH)、6-pyruvoyl-四氢生物喋呤合成酶(6-pyruvoyltetrahydropterinsynthetase,6-PTS)的缺乏。不同国家和地区及不同人群中,PKU的发病率不同。我国在对12个城市的新生儿筛查结果中,平均发病率为1/16500。北方城市较南方城市发病率高,广州地区很低,约为1/10万。
苯丙酮尿症的病因?
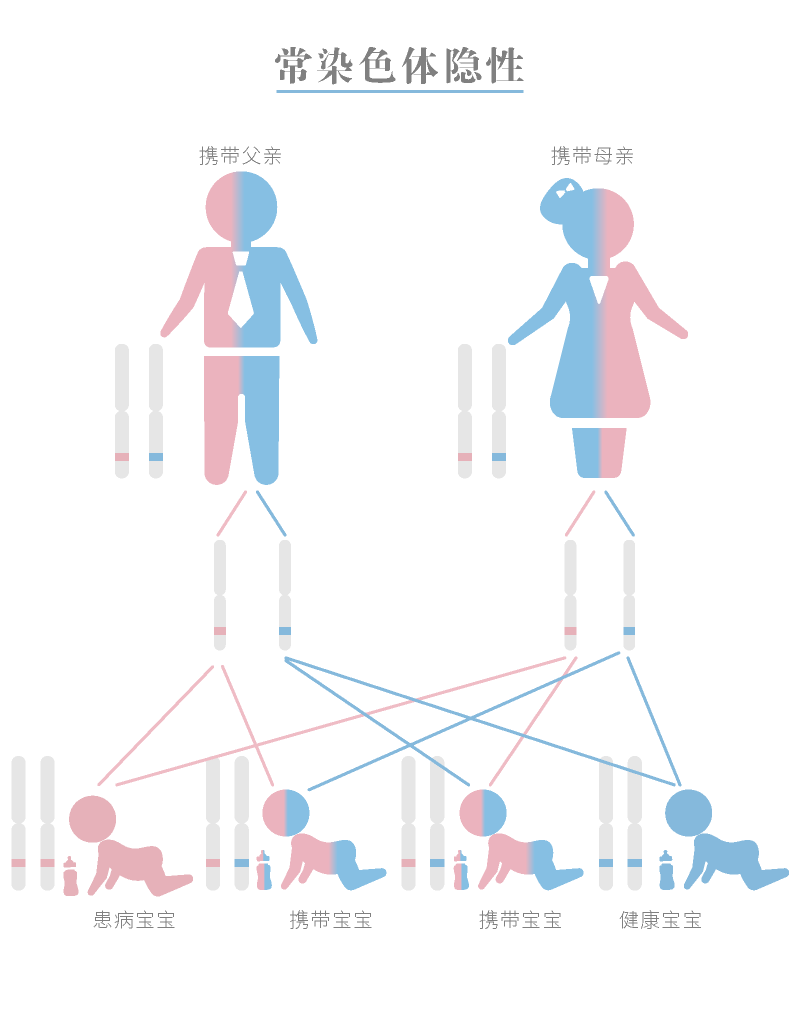
PKU是一种常染色体隐性遗传疾病,意味着您需要在这两个基因拷贝上继承突变来发展这种疾病的症状。携带者没有这种疾病的症状,但可以将有缺陷的基因传给他或她的孩子。如果父母双方携带一份错误基因,他们每个孩子都有四分之一的机会出生。
苯丙酮尿症的症状是什么?
出生苯丙酮尿症的婴儿起初通常没有症状。但如果不治疗疾病,宝宝会受到严重的脑损伤。这种损害可以导致癫痫,行为问题,并阻碍宝宝的成长。其他症状包括湿疹(皮疹),发霉的体味(太多的苯丙氨酸),小头(小头畸形)和白皙的皮肤(因为苯丙氨酸是皮肤色素沉着所必需的)。
如何诊断苯丙酮尿症?
由于苯丙酮尿症PKU必须早期治疗,美国每个州的婴儿都会经常接受这种疾病的检测。从婴儿的脚跟或手臂取出一小块血液样本,并在实验室检查高浓度的苯丙氨酸。
苯丙酮尿症如何治疗?
有PKU的人必须吃低蛋白质饮食,因为几乎所有的蛋白质都含有苯丙氨酸。婴儿给予特殊配方,不含苯丙氨酸。年龄较大的儿童和成年人必须避免食用蛋白质丰富的食物,如肉,蛋,奶酪和坚果。他们还必须避免含有苯丙氨酸的人造甜味剂。

苯丙酮尿症的分类
1.经典型PKU
【临床表现】
PKU患者的主要特征为智力低下,平均IQ为40。临床特征还包括鼠尿气味(因尿液中含苯乙酸之故),轻度色素减低,特殊步态、姿态和坐姿,湿疹,癫痫等。白内障和脑钙化常被忽视。未经饮食治疗患者中罕有智力正常者。因广泛开展的对PKU的新生儿筛查和相应的防治措施,部分病例临床表现相对较轻和不典型,但血清苯丙氨酸浓度仍然高出正常者几倍。其生化特征是PAH酶活性为正常人的5%或以下,血苯丙氨酸浓度升高,但酪氨酸浓度正常。
【诊断与防治】
用HPLC进行血苯丙氨酸(Phe)浓度的测定是诊断PKU的主要方法。正常成人血Phe水平为58±15μmol/L,青少年为60±13μmol/L,儿童为62±18μmol/L,新生儿的正常上限为120μmol/L。未经治疗的经典型PKU患者血Phe浓度可达2.4mmol/L。智商(IQ)测定和MRI脑部影像学检查也是PKU诊断的重要手段。目前应用不同的方法在已知PAH基因突变的家系中,可进行包括携带者在内的基因诊断和产前诊断。新生儿筛查试验目前仍以细菌抑制法(Guthrie试验)为主,采集生后3天新生儿足跟血,滴于滤纸片。筛查阳性者,测定血Phe浓度确诊。
PKU的防治重在早期诊断,及时防治。本病是新生儿筛查疾病中的代表性疾病之一,多数国家已实施。我国许多大中型城市也已开展,有的省市已列入母婴保健法实施细则中,但普及面尚有待提高。防治上主要通过限制苯丙氨酸的摄入,包括素食餐、低动物蛋白、特制奶粉等,可有效减低智力损害,系统治疗的患儿,智力多数正常。新生儿血Phe浓度持续高于360-600μM时,应接受饮食治疗。非经典型PKU单纯用饮食限制疗法难以奏效。因此饮食治疗前,必须明确区分经典型和非经典型。对于患经典型PKU的妇女,可能导致母源性PKU,未接受饮食治疗的经典型PKU的孕妇中,约90%的胎儿会出现智低、小脑、先心、发育不良等损害,称之为母源性PKU。怀孕前接受系统的低Phe限制疗法也可有效地减低对胎儿的损害。对难治性PKU妇女,有建议取夫妇双方配子进行体外受精后,植入替代母亲子宫内妊娠,以避免母源性PKU的危害。一种来自植物的苯丙氨酸氨基裂解酶(PAL)可在肠道中保留较长时间的活性,降解从食物蛋白中获得的Phe,而用于治疗PKU。目前正尝试利用基因工程技术生产药用PAL。肝移植治疗PKU不是常用手段。
【遗传咨询】
PKU严格遵守常染色体隐性遗传的规律。按西方国家平均发病率1/10,000计算,PKU基因频率为0.01,杂合子频率约为0.02。近亲结婚是本病发病率升高的主要原因。许多患者是由于突变等位基因的复合杂合子所引起,而不是由于突变等位基因的纯合子引起。
2.非经典型PKU
非经典型PKU约占PKU的1%,主要因缺乏四氢生物喋呤(BH4)引起。BH4的缺乏是由于二氢生物喋呤(BH2)的生物合成缺陷,或二氢生物喋啶还原酶(DHPR)缺乏后BH4再生障碍所致。BH4是PAH、酪氨酸羟化酶、色氨酸羟化酶等的辅因子,其缺乏除PKU的症状外,也因脑组织中两种神经递质多巴胺和组织胺的减少引起相应的神经系统损害。约60%的生物喋呤代谢障碍是因6-pyruvoyl-四氢生物喋呤合成酶(6-PTS)缺乏所引起,其余多因DHPR酶缺乏所致,也有因GTP环水解酶(GTP-CH)缺乏的报道。
6-PTS缺乏症主要为神经系统受损症状,表现为手臂和大腿肌张力增高、躯干活动受限、易激动、吞咽困难、高热、癫痫发作、震颤、智力发育障碍等。新生儿筛查时发现婴儿高苯丙氨酸血症和尿液、脑脊液异常生物喋呤代谢,部分病例血Phe水平可正常。属常染色体隐性遗传,基因定位于11q22-23,已报道数种基因突变引起的酶缺乏。目前对6-PTS缺乏症尚缺乏有效的治疗,可试用BH4、左旋多巴、5-羟色胺等替代治疗方法,改善症状。
GTP-CH缺乏症症状与6-PTS缺乏症相似,已报道3例病例。属常染色体隐性遗传,基因定位于14q22。测定肝组织GTP-CH酶活性可诊断。口服生物喋呤可用作替代治疗。
DHPR缺乏症约占持续高苯丙氨酸血症患儿的0.5%。DHPR是促进BH4再生的酶,其缺乏可继发PAH、酪氨酸羟化酶、色氨酸羟化酶等酶的缺乏,导致高苯丙氨酸血症、多巴及5-羟色胺缺乏等,叶酸代谢也受影响。其临床损害较严重,表现为进行性神经变性。DHPR基因定位于4p15.3,靠近亨廷顿氏病的基因位点,该基因编码分子量为25,744,由244个氨基酸组成的蛋白质。已鉴定一些碱基置换、插入或缺失突变导致的酶缺乏。通过测定白细胞或红细胞DHPR酶活性可明确诊断。可用干血片法测定酶活性,用作新生儿筛查试验。早期诊断并严格限制饮食中Phe浓度,可取得一定疗效,同时补充5-羟色胺、左旋多巴、叶酸等,补充BH4无疗效,因不能进入BH2的再生循环。

